This document describes how to determine parameters for the
effective Hamiltonian in the feram code http://loto.sourceforge.net/feram/ .
We determine the parameters for BaTiO3 from first-principles calculations.
Theories in background are written in
[Takeshi Nishimatsu, Masaya Iwamoto, Yoshiyuki Kawazoe, and Umesh V. Waghmare:
"First-principles accurate total-energy surfaces for polar structural distortions
of BaTiO3, PbTiO3, and SrTiO3: consequences to structural transition temperatures",
Phys. Rev. B, vol.82, p.134106 (2010) http://dx.doi.org/10.1103/PhysRevB.82.134106 ].
Equations referred in this document are those ones in the PRB article.
You may find latest version of this document in http://loto.sourceforge.net/feram/parameters/ .
You can also find this document and input files in the feram source package, feram-X.YY.ZZ.tar.gz.
% emacs perovskite-B11-12.in # Write a0 for the acell parameter.
% sh perovskite-B11-12.nqs # perovskite-B11-12.dat will be made.
% gnuplot perovskite-B11-12.gp
:
B11-B12 = 81.7753050280732 [eV]
% gv perovskite-B11-12.eps
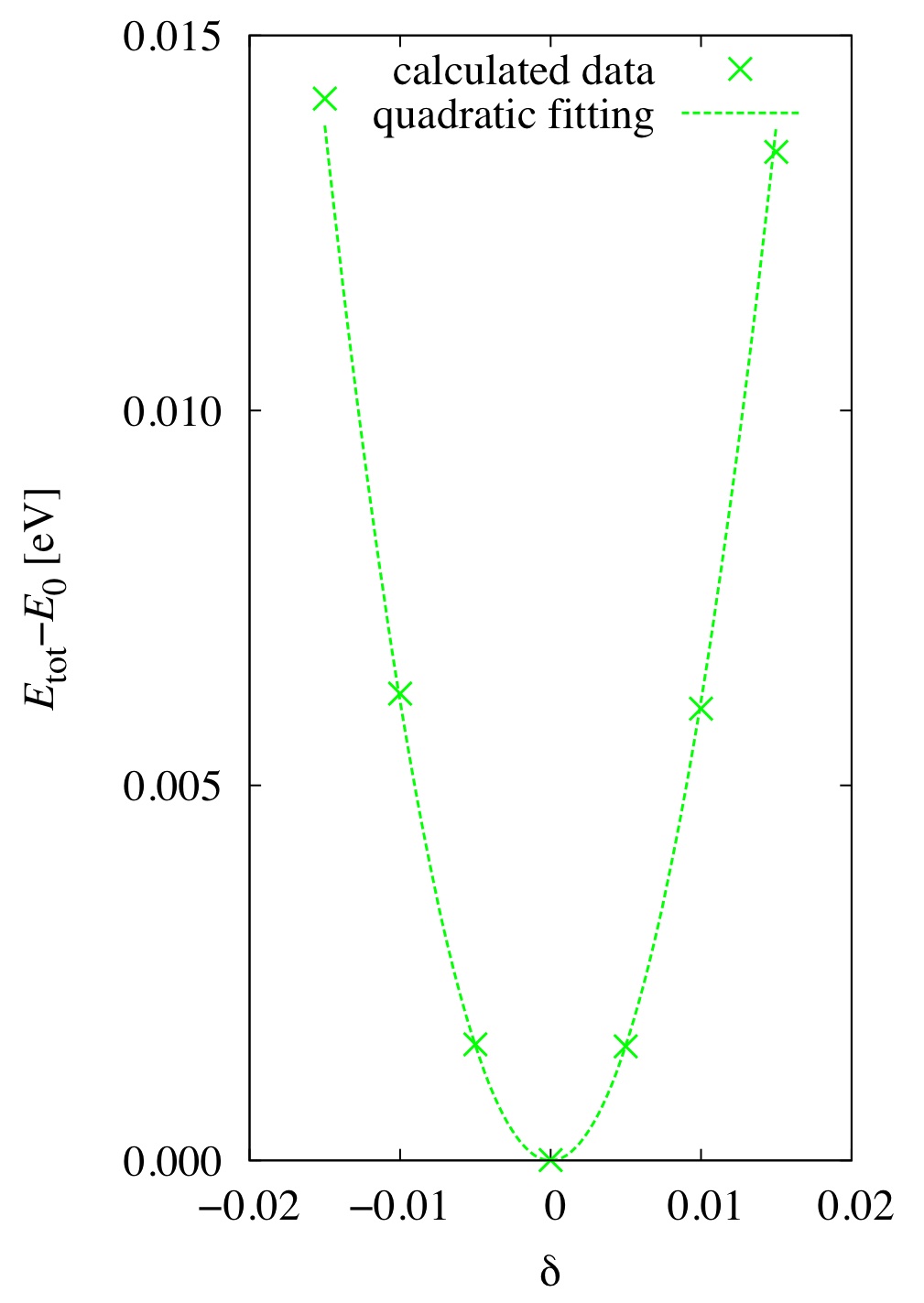
Figure 2: Quadratic fitting of calculated strain dependence of Etot-E_0.
From the centrosymmetric cubic structure, constraint-tetragonal strain is applied.
% emacs perovskite-B44in # Write a0 for the acell parameter.
% sh perovskite-B44.nqs # perovskite-B44.dat will be made.
% gnuplot perovskite-B44.gp
:
B44 = 49.2408864348646 [eV]
% gv perovskite-B44.eps
Figure 3: Quadratic fitting of calculated strain dependence of Etot-E_0.
From the centrosymmetric cubic structure, rhombohedral strain is applied.
The numbers of irreducible k-points for these calculations are
nkpt=40 for B1112 and B11-12 and nkpt=60 for B44.
Polynomial coefficients P_* and coupling constants B1xx, B1yy and B4yz
We determine the potential surface of ABO3 with the method described in [T. Hashimoto,
T. Nishimatsu, H. Mizuseki, Y. Kawazoe, A. Sasaki and Y. Ikeda: Jpn. J. Appl. Phys. 43,
6785-6792 (2004) http://dx.doi.org/10.1143/JJAP.43.6785 ].
Patch for ABINIT
We apply our original patch to ABINIT, rename from abinit to abinit-xyz, then use it.
This brdmin-6.2.3-2011-07-01.patch is applicable to abinit-6.2.3.
% wget http://ftp.abinit.org/abinit-6.2.3.tar.gz
% tar xf abinit-6.2.3.tar.gz
% cd abinit-6.2.3
% mkdir x86_64-Linux-mpif90-gfortran-4.3.3-O3-perovskite-xyz
% cd x86_64-Linux-mpif90-gfortran-4.3.3-O3-perovskite-xyz/
% ../configure FC=mpif90 --enable-mpi --with-mpi-level=2 --disable-netcdf --disable-libxc --disable-etsf-io
% cd src/95_drive/
% cp ../../../src/21drive/brdmin.F90 .
% cp ../../../src/21drive/brdmin_init.F90 .
% cp ../../../src/21drive/interfaces_95_drive.F90 .
% patch -p0 < SOMEWERE/brdmin-6.2.3-2011-07-01.patch
% cd ../..
% make
% cd src/main
% mv abinit abinit-xyz
Input file generator
Generate input files with each ruby script in the directory BaTiO3-WuCohenGGA.
% sh perovskite-optcell2-001.nqs # results in perovskite-optcell2-001.dat
% sh perovskite-optcell2-110.nqs # results in perovskite-optcell2-110.dat
% sh perovskite-optcell2-111.nqs # results in perovskite-optcell2-111.dat
B1xx, B1yy, B4yz, P_k1, P_k2, P_k3, P_k4, P_alpha and P_gamma
It is quite difficult to express the total-energy surfaces
even with up to 8th order polynomial in wide range of u.
Therefore, we fit Eqs. (14a)--(14c) only
to the calculated data points within narrow range of u.
E0, a0, B11, B12, and B44 must be written in BaTiO3-WuCohenGGA/perovskite-optcell2.gp.
% emacs perovskite-optcell2-001-narrow.dat # narrow range of u of perovskite-optcell2-001.dat
% emacs perovskite-optcell2-110-narrow.dat # narrow range of u of perovskite-optcell2-110.dat
% emacs perovskite-optcell2-111-narrow.dat # narrow range of u of perovskite-optcell2-111.dat
% ./perovskite-optcell2.gp
:
B1xx = -185.347187551195 [eV/Angstrom^2]
B1yy = -3.28092949275452 [eV/Angstrom^2]
B4yz = -14.5501738943852 [eV/Angstrom^2]
P_k1 = -267.980139917128 [eV/Angstrom^6]
P_k2 = 197.500718362569 [eV/Angstrom^6]
P_k3 = 830.19997929324 [eV/Angstrom^6]
P_k4 = 641.968099408291 [eV/Angstrom^8]
P_alpha = 78.9866142426711 [eV/Angstrom^4]
P_gamma = -115.484148812671 [eV/Angstrom^4]
#kappa = -1.51821042113559 [eV/Angstrom^2] <=== We will also use this value to determine P_kappa2.
% gv perovskite-optcell2.eps
Figure 4: GNUPLOT drawing to determine polynomial coefficients P_* and coupling constants B1xx, B1yy and B4yz.
Filled points are selected data in narrow ranges of u.
Eigenvalues and eigenvectors of IFC matrix at the Gamma point
Eigenvalues and eigenvectors of IFC matrix at the Gamma point
can be calculated with frozen phonon calculations at the Gamma.
The program feram-X.YY.ZZ/src/feram_frozen_phonon_Gamma.F help us to
do it. These eigenvalues and eigenvectors must be similar to those
of calculated from following response-function calculations.
% sh perovskite-frozen-phonon-Gamma.nqs # perovskite-frozen-phonon-Gamma.dat will be made.
% feram-X.YY.ZZ/src/feram_frozen_phonon_Gamma # this program reads perovskite-frozen-phonon-Gamma.dat
a0 = 3.9859580 [Angstrom]
eigenvalues [eV/Angstrom^2] and eigenvectors of force_constant_matrix
1 -3.982367 0.1648 0.7726 -0.2003 -0.2003 -0.5438 <=== Gamma_15 soft mode
2 0.058552 0.4511 0.4482 0.4458 0.4458 0.4451
3 4.682338 -0.0000 0.0000 0.7071 -0.7071 0.0000
4 8.125787 0.8555 -0.3118 -0.2915 -0.2915 0.0309
5 13.605841 -0.1936 0.3240 -0.4197 -0.4197 0.7108
Response-function calculations
We perform some response-function calculations with ABINIT (RF calculations,
See http://www.abinit.org/documentation/helpfiles/for-v6.8/tutorial/lesson_rf1.html and
[Xavier Gonze and Chngyol Lee: Phys. Rev B vol.55, pp.10355-10368 (1997)
http://dx.doi.org/10.1103/PhysRevB.55.10355 ]) to determine optical dielectric
constant epsilon_inf, effective charge Z_star, effective mass mass_amu,
self interaction P_kappa2, and short range interactions j1, ..., j7.
We do NOT move ions explicitly.
Using feram-X.YY.ZZ/src/feram_diagonalize15x15.F,
eigenvalues and eigenvectors of IFC matrices
of each k-point are calculated. Dashed lines
in Fig. 5 (B) is a plot of the eigenvalues of
IFC matrices along symmetric axes in the
first Brillouin zone.
How to plot eigenvalues of IFC matrices is described in
http://forum.abinit.org/viewtopic.php?f=12&t=1273 .
% less perovskite-Gamma.out
:
Dielectric tensor, in cartesian coordinates,
j1 j2 matrix element
dir pert dir pert real part imaginary part
1 7 1 7 6.8691464565 0.0000000000
1 7 2 7 0.0000000000 0.0000000000
1 7 3 7 0.0000000000 0.0000000000
2 7 1 7 0.0000000000 0.0000000000
2 7 2 7 6.8691464565 0.0000000000
2 7 3 7 0.0000000000 0.0000000000
3 7 1 7 0.0000000000 0.0000000000
3 7 2 7 0.0000000000 0.0000000000
3 7 3 7 6.8691464565 0.0000000000
:
Effective charge Z_star and effective mass mass_amu
We can find the calculated Born effective charges for each atom in the
file of BaTiO3-WuCohenGGA/force-constant-matrix/perovskite-Gamma.out.
There are two ways of calculations of effective charges,
(from electric field response) and (from phonon response).
They must be identical within some error.
% less perovskite-Gamma.out
:
Effective charges, in cartesian coordinates,
(from electric field response)
if specified in the inputs, asr has been imposed
j1 j2 matrix element
dir pert dir pert real part imaginary part
1 1 1 7 2.7419582957 0.0000000000
2 1 1 7 0.0000000000 0.0000000000
3 1 1 7 0.0000000000 0.0000000000
1 2 1 7 7.4934093087 0.0000000000
2 2 1 7 0.0000000000 0.0000000000
3 2 1 7 0.0000000000 0.0000000000
1 3 1 7 -5.9318277473 0.0000000000
2 3 1 7 0.0000000000 0.0000000000
3 3 1 7 0.0000000000 0.0000000000
1 4 1 7 -2.1492074350 0.0000000000
2 4 1 7 0.0000000000 0.0000000000
3 4 1 7 0.0000000000 0.0000000000
1 5 1 7 -2.1492081943 0.0000000000
2 5 1 7 0.0000000000 0.0000000000
3 5 1 7 0.0000000000 0.0000000000
:
The direction of the Gamma_15 soft mode and
The direction to the minimum are not same in principle.
Using one of them, we determine the effective charge
Z_star and the effective mass mass_amu.
Figure 5: (A) Half of eigenvalues of the 3x3 long-range dipole-dipole interaction matrix Phi(k)
are plotted along symmetric axes in the first Brillouin zone of the simple-cubic lattice.
Special points and k/(2pi)=(1/4, 1/4, 0) (the center of the Sigma axis) are
indicated with vertical dotted lines. Labels (a)--(g)
corresponds to Eqs. (15a)--(15g), respectively.
Tics in the unit of Z^2/epsilon/a^3 is placed in left side.
Tics in the unit of eV, in the case of the parameter set of [Takeshi Nishimatsu,
Masaya Iwamoto, Yoshiyuki Kawazoe, and Umesh V. Waghmare: Phys. Rev. B 82, 134106 (2010)]
is placed in right side.
(B) Half of eigenvalues of the calculated 15x15 inter-atomic force constant (IFC) matrix (dashed black lines).
Half of eigenvalues of the total (long-range + short-range) interaction matrix Phi^quad(k) (solid red line).
Difference between them is the elevation of 0.38795.